Гарвардські науковці модифікували метод редагування геному CRISPR у такий спосіб, щоб пристосувати його до лікування дитячої прогерії. Піддослідні миші з синдромом Гетчінсона-Гілфорда, на яких випробували метод, прожили вдвічі довше, ніж неліковані тварини, а також мали краще здоров’я та були активнішими. Такі результати описані в статті журналу Nature.
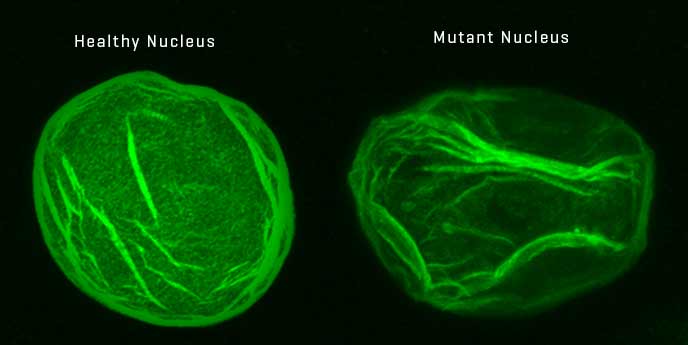
Ядро клітини здорової людини (зліва) та хворої на прогерію (справа). Dr. Susan Parkhurst / Current Biology, 2015
Що це за хвороба?
Прогерія є дуже рідкісним генетичним захворюванням, якого у світі зареєстровано всього лише близько 400 випадків. Хвороба характеризується передчасним старінням організму, через що пацієнти з дитячою прогерією, або синдромом Гетчінсона-Гілфорда, живуть в середньому 14 років. Стан спричиняється мутацією всього однієї “букви” ДНК в одній з двох копій гену, що кодує білок ламін А. Дефектний ген призводить до вироблення організмом такого ж дефектного білка, що має назву прогерин. Саме він перешкоджає нормальному поділу клітин та викликає симптоми раннього старіння.
Генна терапія може допомогти таким хворим?
Звичайні методи генної терапії, які полягають у додаванні до геному правильного варіанту гену, у цьому випадку не спрацюють, оскільки пацієнт вже має одну правильну версію гена. Проблема полягає саме у наявності хибної версії. Стандартний метод редагування геному CRISPR, який ще називають генетичними ножицями, потенційно міг би допомогти. Він може розрізати ДНК у потрібному місці, однак в такому разі є ризик пошкодити й здорову версію гену, що теж погіршує стан хворих.
Вчені з Гарвардського університету та Інституту Брода запропонували інший підхід. Вони модифікували CRISPR так, щоб він розрізав не обидві нитки ДНК, а лише одну з них, і замінював помилкову основу на правильну.
Наскільки це дієво?
Вчені випробували метод спочатку на культурі людських клітин від двох пацієнтів з прогерією. Коли спроба виправити мутацію виявилася успішною, перейшли на мишей із людським варіантом мутації білка ламін А. Для цього науковці ввели хворим піддослідним мишам ін’єкцію з адено-асоційованими вірусами, що доставили інструмент редагування геному. На той час мишам було всього два тижні, що еквівалентно п’ятирічному віку в людини. Із прогерією вони мали прожити близько 215 днів.
Через пів року після ін’єкції від 20 до 60 відсотків клітин кісток, м’язів, печінки, серця та аорти піддослідних мишей містили в собі виправлений варіант гена. Рівень прогерину суттєво знизився, а ламіну А, навпаки, збільшився в багатьох тканинах. Ці тварини були набагато активнішими, ніж неліковані, та врешті прожили вдвічі довше — у середньому 510 днів.
Автори роботи сподіваються, що за умови успішних подальших дослідів для кращих результатів їхній винахід можна буде поєднати із ліками проти прогерії, перші з яких американський регулятор схвалив у листопаді минулого року. Цей препарат дає змогу подовжити життя пацієнтів із синдромом Гетчінсона-Гілфорда у середньому на 2,5 роки.
Як редагують геном та чого це потрібно, читайте в нашому розборі "Що таке геном? Для чого його редагувати?".



